Abstract
Non-contact anterior cruciate ligament (ACL) rupture is a common serious orthopaedic disease in humans and dogs. Familial risk has been recognized in both species but interactions between genetic effects and environmental risk are not understood. We investigated ACL rupture heritability, genetic architecture, selection pressure, sharing of risk genes and biological pathways, and polygenic risk score (PRS) prediction of disease risk. In both species, ACL rupture has moderate heritability, is likely under negative selection, and has a highly polygenic architecture where thousands of variant effects act together to influence disease risk. In dogs, we found hotspots of regional heritability. We also confirmed sharing of multiple risk genes. Our findings challenge the dogma that non-contact ACL rupture is predominantly due to a single overload injury event. Our results also suggest that accurate PRS prediction of ACL rupture risk is an achievable goal in both species, enabling identification of individuals for personalized medical care.
Introduction
Polygenic risk score (PRS) prediction has provided a valuable tool for assessing an individual’s genetic predisposition to a given complex disease1. While PRS prediction has shown promise for early disease detection, precision medicine, and genetic counseling, its application to human musculoskeletal disease is limited, in contrast to cardiovascular disease, diabetes, and psychiatric disorders2,3,4. By leveraging genetic information alongside other risk factors like age and family history, PRS prediction has the potential to enhance clinical screening for disease risk and enable early intervention for the treatment of orthopaedic disease and improved management of athletes in sports medicine. Genetic studies in the spontaneous dog model5 can be used to enrich human genome-wide association study (GWAS) by focusing on cross species shared genes and pathways, thereby accelerating progress with PRS studies of orthopaedic disease.
Non-contact anterior cruciate ligament (ACL) rupture is an economically important common heritable spontaneous disease with serious long-term sequelae in both humans and dogs, with high incident rates in both species5,6,7,8,9. Anatomic features include comparable intra-articular structures (5, and Fig. 1). There is overwhelming evidence that a fatigue mechanism with progressive ligament fiber rupture explains most cases of non-contact ACL rupture in both species5,10,11 with frequent second contralateral ruptures12,13. Up to 50% of human cases develop moderate-to-severe posttraumatic osteoarthritis14,15. Intrinsic/extrinsic risk has been extensively investigated in humans16,17, but PRS prediction has not been studied. The current dogma that non-contact ACL tears occur principally because of trauma from a single loading cycle that exceeds ACL ultimate tensile strength (one cycle on x axis above ultimate tensile strength on y axis in Fig. 2) must be updated18. Adaptive responses in the ligament19 are likely under genetic control. While single-event overload ruptures do occur, increasing evidence supports the concept that ligament failure often occurs because of fatigue damage from repetitive loading. In this regard, a small ACL cross-sectional area appears important. In dogs, it has been widely documented that traumatic ACL ruptures are rare, and the vast majority represent non-contact ruptures resulting from fatigue injury11.

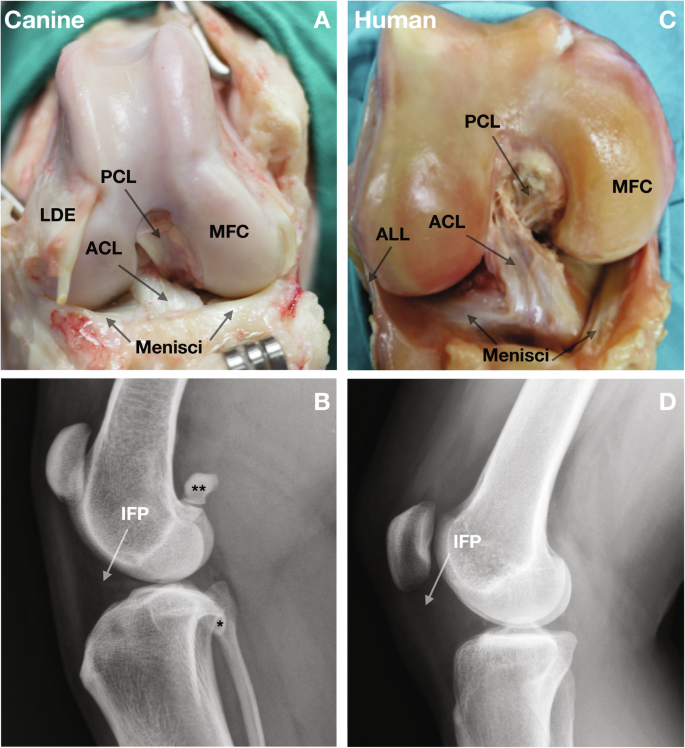
Anatomic and radiographic features of the dog (A, B) and human knee (C, D). The similarity is obvious between the two species, but dogs do not have an ALL. ACL anterior cruciate ligament, PCL posterior cruciate ligament, MFC medial femoral condyle, LDE long digital extensor, ALL anterolateral ligament, IFP infrapatellar fat pad. The canine knee includes sesamoid bones in the tendon of origin of the lateral and medial gastrocnemius (**), and the popliteal tendon (*).

A Theoretical stress-life plot and patterns of fatigue failure risk for different combinations of loading magnitude and loading cycles. where A is a proportionality constant, and b is the slope of the S-N curve. Fatigue life is defined as the number of cycles to failure Nf at a particular stress magnitude σ. B Illustrating the inverse relationship between force applied to the anterior cruciate ligament (ACL) and the number of near-maximal loading cycles it can withstand before failing [adapted from (https://doi.org/10.1080/00140139.2016.1208848), A, and ref. 10 B]. Copyright © Taylor & Francis Group. Revised and used with permission.
Complex epidemiological features such as breed predisposition9, ligament matrix degeneration11, obesity20, conformation20, and joint immune responses11 promote ligament weakening and fatigue injury in dogs. Such observations are consistent with the hypothesis that non-contact ACL rupture is a polygenic complex disease with genetic and environmental risk where subjects with non-contact ACL rupture carry an elevated portfolio of genetic risk variants associated with considerable clinical and genetic heterogeneity21. Familial risk of non-contact ACL rupture has been found in both species9,22. Heritability estimates range from 0.27 to 0.85 in dogs and 0.69 in humans7,23,24,25,26. Dog discovery GWAS suggested a highly polygenic genetic architecture23. Candidate genes identified through GWAS have small effect sizes, supporting the hypothesis that the genetic risk of non-contact ACL rupture is highly polygenic27.
PRS using a risk allele counting method showed promise as a classifier for risk prediction of non-contact ACL rupture in the Labrador Retriever23,28. The inclusion of non-genetic risk factors (covariates) in the models improved prediction accuracy using single-nucleotide polymorphism (SNP) markers29. The number of genetic variants influencing non-contact ACL rupture risk in humans and dogs remains unclear. Variants from highly powered GWAS show stronger pathway enrichment and could explain a substantial proportion of trait heritability30. Influential variants are often clustered together in a hotspot in the genome30. Accurate prediction of a polygenic human phenotype, such as height, is possible with ~100,000 individuals31. GWAS of a purebred dog population is advantageous because linkage disequilibrium (LD) in dogs is up to ~100-fold more than human populations32. This suggests a reasonable starting point for robust analysis of a polygenic disease is ~1000 dogs. The purpose of this study was to generate a large reference population of Labrador Retrievers phenotyped as non-contact ACL rupture cases or controls to obtain definitive estimates of heritability, genetic architecture, and to undertake PRS prediction in dogs using this reference population as a training set. We also undertook similar analyses for humans using GWAS summary statistics. We confirmed moderate SNP heritability for non-contact ACL rupture in the Labrador Retriever and a highly polygenic architecture and showed that PRS prediction is a promising approach for predicting risk of non-contact ACL rupture in dogs with prediction accuracy approaching 70%. Similar results were obtained for human ACL rupture. Such findings advance the concept of using genetic investigation of the spontaneous orthopaedic disease in the dog model to advance and enrich studies of human orthopaedic disease, such as establishing a computational framework capable of quantifying the genome-wide genetic liability associated with the risk of human non-contact ACL rupture for injury prevention screening.
Results
Heritability of non-contact ACL rupture in dogs and humans
Dog ACL rupture heritability was estimated as h2o = 0.57 ± 0.06 and h2l = 0.52 ± 0.06 on the observed and continuous liability scales, respectively, using the restricted maximum likelihood (REML) approach. The probit Bayesian linear mixed model yielded a SNP-based heritability estimate of h2l = 0.63 ± 0.08 on the liability scale (Fig. S1). In addition, the Bayesian-based models yielded dog ACL rupture SNP-based heritability estimates of h2l = 0.52 ± 0.05 and h2l = 0.50 ± 0.097 for BayesR and BayesS, respectively on the liability scale (Fig. 3). In humans, summary GWAS statistics SNP-based heritability estimates were h2l = 0.30 ± 0.08 and h2l = 0.33 ± 0.06 for sBayesS and sBayesR, respectively (Fig. 4). SNP-based heritability of human ACL rupture was estimated at h2l = 0.23 ± 0.084 using LD score regression. Given these estimates of heritability, we next estimated its regional distribution across the genome.

A The proportion of single-nucleotide polymorphisms (SNPs) with small (10−4 × σ2g), medium (10−3 × σ2g), and large (10−2 × σ2g) effects on the trait was estimated across the genome using BayesR. Large effect SNPs were enriched in chromosomes 1, 15, 29, and 35 where there is also enhanced regional heritability (Fig. 5). Risk SNPs were present across the entire genome. B We found heritability on the continuous liability scale was h2l = 0.52 ± 0.05 and h2l = 0.50 ± 0.097 for BayesR and BayesS, respectively. The posterior distribution of the BayesS S hyperparameter is also depicted. We found S = -0.56 ± 0.45, indicating negative selection pressure on non-contact ACL rupture in dogs. The red dashed lines represent the highest posterior density (HPD) 95% confidence intervals and the gray dashed line “represents the mean” for each distribution. C There were 5.19% non-null and 94.81% null SNPs that influence the risk of non-contact ACL rupture with BayesS analysis. BayesR indicated of 0.01% large, 0.29% medium, and 3.41% small effect SNPs, together with 96.29% null SNPs. n = 1006 biologically independent Labrador Retriever dogs.

The proportion of single-nucleotide polymorphisms (SNPs) with large (10−2 × σ2g), medium (10−3 × σ2g) and small effects (10−4 × σ2g) on the trait was estimated using sBayesR. A SNP heritability was estimated at 0.3 and 0.33 using sBayesS and sBayesR, respectively. The posterior distribution of the sBayesS hyperparameter is also depicted. We found S = −0.59 ± 0.36, again indicating negative selection pressure on risk of ACL rupture. The red dashed lines represent the highest posterior density (HPD) 95% confidence intervals and the gray dashed line “represents the mean” for each distribution. B Using sBayesS, we found that 3.44% of SNPs had estimable effects on non-contact ACL rupture risk. Using sBayesR, these SNPs consisted of 0.82% large, 5.93% medium, and 1.47% small effect SNPs, together with 91.78% null SNPs.
Regional heritability of dog non-contact ACL rupture
By partitioning the genome using the genetic variant distributions, we used 4753 genomic windows for this analysis with 30 SNPs per window. We captured the variance of each 0.484 ± 0.076 cM segment of the genome (Fig. S2). Chromosome 1 and X had the largest number of windows with 245 and 136 windows, respectively. Heritability hotspots were evident in several regions of the genome, particularly chromosomes 1, 4, 9, and 15 (Fig. S3). Given evidence of hotspots of enriched regional heritability, we further investigated the genetic architecture of non-contact ACL rupture and trait selection pressure using a Bayesian analytical approach.
Non-contact ACL rupture genetic architecture and selection pressure
The proportion of phenotypic variance explained by SNPs with different effect sizes on each chromosome in dogs is depicted in Fig. 3. Thousands of non-null SNPs were identified using BayesR with 96.29%, 3.41%, 0.29%, and 0.01% SNPs assigned to the four effect size classes: null, small, medium, or large (Fig. 3). Using BayesS, which examines the relationship between SNP effect size and minor allele frequency (MAF), we found non-contact ACL rupture is under negative selection in dogs ((hat{S}) = −0.56 ± 0.45) and ~87% of the posterior samples for S value were negative. BayesS showed 5.19% of non-null and 94.81% null SNPs, like BayesR (Figs. 3 and S4).
In humans, we found that 3.44% of SNPs had estimable effects on ACL rupture risk using sBayesS with 96.56% null SNP (Fig. 4). We found human non-contact ACL rupture is also under negative selection ((hat{S}) = −0.59 ± 0.36), and ~97% of the posterior samples were negative (Figs. 4 and S4). We also found 91.78%, 1.47%, 5.93%, and 0.82% of SNPs were assigned to the null, small, medium, and large effect mixture classes using the sBayesR model.
Cross-species pleiotropy analysis for risk of ACL rupture
Given the findings that both canine and human non-contact ACL rupture has a polygenic genetic architecture, we performed an analysis of GWAS summary statistics for shared risk genes by mapping the closest genes to the GWAS candidate loci. We identified 16 non-contact ACL rupture risk genes shared between humans and dogs, including the protein tyrosine kinase receptors PTPRT and PTPRM as depicted in Fig. 5. Several of the shared risk genes related to skeletal muscle homeostasis (Supplementary Results). Risk SNPs/genes were clustered in hotspots in both species. A comprehensive analysis of canine and human ACL rupture risk genes from published literature is represented in Figs. S5 and S6, of which there are 50 shared genes. Biological pathway analysis of non-contact ACL rupture risk genes showed the most significant pathway was extracellular matrix organization in both species (Figs. S7–S9 and Supplementary Results). Given evidence of shared risk genes influencing the development of a heritable complex disease, we investigated the accuracy of non-contact ACL rupture PRS risk prediction.

In a cross-species pleiotropy analysis, 16 shared genes influencing risk of non-contact ACL rupture were identified. Fisher’s Exact test was used to determine if whether the16 shared genes are significantly higher than sampling genes at random by chance. The P value from Fisher’s Exact Test was P = 0.0083. Sharing was significantly beyond what would be expected by chance, implying meaningful biological connections between the two species. Hotspots within the genome were found in both species, particularly human chromosome 16 and dog chromosome 5. n = 1006 biologically independent Labrador Retriever dogs.
Polygenic risk score prediction of non-contact ACL rupture risk
We estimated the predictive performance of Bayesian and machine learning models for non-contact ACL rupture risk prediction in dogs. GWAS was performed for SNP selection further confirming trait polygenicity (Fig. S10). Results of the ten-fold cross-validation GWAS using model-specific top GWAS SNPs are represented in Fig. 6. To select the optimum number of SNPs for fitting the prediction models, we conducted a grid search and evaluated the model across different percentages of SNP sets. The weighted subspace random forest (RF), support vector machine (SVM), least absolute shrinkage and selection operator (LASSO), and elastic net (EN) models showed the best performance with 1% (1420 SNPs), 7% (9944 SNPs), 2% (2841 SNPs), and 2% (2841 SNPs) of total SNPs, respectively. In addition, all Bayesian regression models had the best performance when we selected the top 30% of SNPs (Fig. S11). Several approaches were used to evaluate metrics used to measure the predictive ability of models including area under the curve (AUC) (Fig. S12). As represented in the Fig. 6, Bayesian Lasso (BL) exhibited the highest AUC mean of 0.695±0.036, closely followed by Bayesian Ridge Regression (BRR), Bayes C (BC) and Bayes B (BB). Conversely, the RF model displayed the lowest AUC mean (0.590 ± 0.0479), indicating a less robust ability to distinguish between outcomes. Bayesian models outperformed machine learning models with an average AUC of 0.683 ± 0.147 versus 0.616 ± 0.141. The R2 on the liability scale metric reflects the proportion of the explained variance in the case-control outcomes by each model. In this regard, BL yielded the highest R2 (0.362 ± 0.116), denoting the model’s effectiveness in explaining the variance. The RF model had the lowest R2 (0.259 ± 0.134). Again, Bayesian regression models outperformed machine learning models with a slightly higher average R2 (0.333 ± 0.148) for predicting non-contact ACL rupture risk compared to (0.296 ± 0.141). Again, BL presented the highest accuracy (0.647 ± 0.0649). The average F1 score for Bayesian regression models was 0.472 ± 0.0014, marginally higher than that for machine learning models at 0.468 ± 0.012. RF had the highest F1 score (0.487 ± 0.055), and EN had the lowest (0.454 ± 0.051). When overall PRS prediction in dogs and humans was evaluated, despite similar SNP-based heritability, the coefficient of determination for human non-contact ACL rupture was lower than for dogs (Fig. S13).

A We performed ten-fold cross-validation genome-wide association study and studied four machine learning classification models (EN elastic net, LASSO least absolute shrinkage and selection operator, RF weighted subspace random forest, and SVM Support Vector Machine). B Using a similar approach, we also studied four Bayesian regression models (BL Bayesian Lasso, BRR Bayesian Ridge Regression, BB Bayes B, BC Bayes C). We calculated accuracy (ACC), mean area under the curve (AUC), F1 score, and R2 to estimate model predictive performance. The Bayesian models yielded the best predictive accuracy. Bar chart plots represent mean±standard error. Data scatter is also represented. n = 1006 biologically independent Labrador Retriever dogs.
ACL rupture PRS prediction in a multi-ancestry dog population
When the Labrador Retriever reference population was used to predict Rottweiler ACL rupture case-control status, the AUC dropped to 0.616 ± 0.067 from 0.659 ± 0.068 (Fig. 7 and Table S1). By adding 60% of the Rottweilers to the Labrador reference population in each fold of cross-validation and creating a multi-ancestry population, the accuracy was considerably increased to 0.65 ± 0.083. We observed the same pattern for other evaluation metrics, for example the mean accuracy metric LAB → LAB was 0.614 ± 0.065, which was higher than in the LAB → ROT scenario (0.523 ± 0.107). Interestingly, the multi-ancestry scenario yielded an accuracy like the within-breed LAB → LAB prediction (0.594 ± 0.111). The mean F1 score was 0.478 ± 0.041, 0.308 ± 0.047, and 0.367 ± 0.061, respectively, for within-breed, across-breed, and multi-ancestry scenarios. The R2 mean on the liability scale was 0.305 ± 0.134, 0.225 ± 0.024, and 0.345 ± 0.129, respectively, for within-breed, across-breed, and multi-ancestry scenarios.

A The lowest predictive ability metrics were obtained when we used Lab as the reference population to predict PRS in the ROT population (LAB→ROT). When we added Rottweiler dogs (n = 65) to the reference population of 1006 Labrador Retrievers to make a MA training set, the ACL rupture prediction accuracy metrics increased dramatically (MA→ROT). B The linkage disequilibrium (LD) decay plots highlight genomic differences in the two ancestral populations. C The principal components (PC) results which indicates genetic differentiation of two breeds, and D The proportion of variance explained by the first top 10 principal components. Box and whisker plot in (A) represents median, quartiles, range, and outliers. n = 1006 biologically independent Labrador Retriever dogs and 108 Rottweiler dogs.
Discussion
Non-contact ACL rupture is a common economically important disease in humans and dogs33,34, but a shared genetic etiology has not been previously investigated. The condition cannot be fully explained by the biomechanical mechanism of single overload injury to the knee in both dogs and humans. In humans, it remains unclear whether non-contact ACL rupture should be considered an injury or a disease6,10,16,17,18. In human complex trait GWAS, only a fraction of the genetic variants involved in disease have been discovered in past GWAS research, a phenomenon referred to as missing heritability35. Epistasis, de novo mutations, or epigenetic effects may explain some missing heritability21,35. The contribution of multiple rare variants to population genetic variance is unclear because association studies are often underpowered to detect rare variants (< 1% MAF). Such variants may also escape detection by SNP-based GWAS because strong LD with common SNPs is unlikely. Copy number variants may also interfere with the ability to detect adjacent SNPs that are in strong LD and likely have important effects in non-contact ACL rupture35,36,37. Inaccurate phenotyping will affect the average effect sizes across groups and may also cause distinct causal variants to be combined21,35.
Our results confirm that the scientific community should view human non-contact ACL rupture as a heritable disease7 with a polygenic genetic contribution that includes risk genes that are shared with spontaneous dog ACL rupture. Canine genomics, particularly studies of complex traits and diseases, has advanced rapidly from map construction in 200538. Over the past ~200 years, selection for breed creation has caused breed-associated disease in a species with large LD blocks39. The risk of non-contact ACL rupture is higher in specific ancestral populations (breeds)9. This suggests ancient alleles influenced by selection pressure for different physical conformations affect non-contact canine ACL rupture risk, and that risk alleles are likely shared across high-risk breeds. Recent dog non-contact ACL rupture GWAS analyses have contributed to knowledge of disease-associated pathways by pursuing canine GWAS with larger sample sizes, dense numbers of SNPs, stringent phenotyping, and innovative approaches such as multivariate, Bayesian, or categorical GWAS23,27,37,40,41,42. Such data are rare. No one has ever undertaken concurrent analysis of dogs and humans to examine sharing of risk genes, and this approach is a key innovation.
Our findings are particularly noteworthy given the prevailing clinical and scientific view of human non-contact ACL rupture as an injury. Our SNP-based estimates of dog non-contact ACL rupture heritability are like the high value of 0.69 described in a recent human twin study7. An important difference between humans and dogs for SNP-based estimates of heritability is the degree of relatedness in dogs compared with humans, which could help to explain differences in the estimates between the two species. SNP-based estimates of human ACL rupture heritability were also relatively high at 0.30–0.33 based on our analysis of GWAS summary data. Variation in heritability estimates between models likely reflects sample size, model assumptions, and sensitivity to causal variant distribution across the genome. Moderate heritability implies a robust genetic contribution to disease risk that can be captured in PRS values used to predict risk of disease. An improved understanding of complex disease polygenicity in dogs is needed, as it is not widely recognized that common diseases shared between species have a similar polygenic genetic architecture. Past work has suggested that relatively few genes may explain morphologic differences between breeds, based on analysis of average breed size43,44. This past dog research did not study individual variation in complex phenotypes within a breed as was done in the present study. An additive polygenic disease architecture provides a homogenous representation of disease for a dog population that is consistent with a high degree of non-additivity at the biological level and between-subject genetic heterogeneity that is difficult to identify clinically45.
Our analysis also identified hotspots within the genome with enriched regional heritability, suggesting that there are potential clusters of functional variants that influence non-contact ACL rupture risk within the genome46, many of which likely include regulatory SNPs that influence gene expression47. To further investigate, we studied genetic architecture with the BayesR model (SI Appendix, Supplementary Methods), which appropriately models the effect size distribution expected for a complex disease. We confirmed the highly polygenic architecture of non-contact ACL rupture where SNP effects predominantly have small effects on variance in both species, with risk SNPs distributed widely across the genome in the dog. The variance explained by each chromosome was largely related to chromosome length. For example, chromosome 1 and X, the largest chromosomes on the dog genome, respectively, at 120.9 Mb and 124.9 Mb, contributed more genetic variance explained by medium and small effect size variants. In contrast, genetic variance on chromosome 35, which is the second smallest chromosome at 26.3 Mb had a similar genetic variance to chromosome 16 which is more than double the size at 55.4 Mb. In fact, chromosomes 15, 23, 29, and 35 all showed a larger genetic variance due to large effect SNPs and to a lesser extent to SNPs with smaller effects. Additionally, we analyzed SNPs with BayesS (SI Appendix, Supplementary Methods). Our results suggest that mutations with deleterious effects on ACL homeostasis are kept at low frequencies by moderate negative selection in both species, resulting in a negative relationship between effect size and MAF for this disease. Negative selection pressure is like other common human polygenic diseases48. This type of selection pressure on complex diseases is little studied in dogs.
Because of the similarity in the architecture of the genetic contribution to non-contact ACL rupture in humans and dogs, we also estimated cross-species pleiotropy and confirmed shared risk genes, clustered in hotspots across the genome, that are involved in a range of biological processes. Recent human GWAS results49,50 have not identified many strong SNP associations, a result that is likely a consequence of a highly polygenic genetic architecture, based on our findings. We found shared risk genes included two protein tyrosine kinase receptors (PTPRT, PTPRM). ERK1/2 signaling is downregulated by PTPRM51 and JAK-STAT3 signaling is upregulated by PTPRT 52. Both ERK1/2 and STAT3 are expressed in human ruptured ACL53,54 but little is known about the role of these pathways in non-contact ACL rupture pathogenesis. Identification of shared ACL rupture risk genes confirms cross-species pleiotropy for a common polygenic disease and further suggests that dog spontaneous ACL rupture is a suitable translational model for studies of non-contact ACL rupture genetics. These findings provide an important basis for future work where discovery data in the dog can be used to enrich future human GWAS and PRS prediction of ACL rupture risk and shed light on which risk genes might be particularly impactful to disease. Interestingly, none of the shared risk genes were identified as biological priors in an earlier Bayesian GWAS in dogs27. Several of the risk genes relate to skeletal muscle homeostasis, suggesting genetic effects on neuromuscular control may be an underappreciated pathway leading to decreased joint stability55, increased risk of fatigue injury in the ACL, and eventual ligament rupture.
Moderate-to-high heritability of non-contact ACL rupture suggests accurate PRS prediction should be feasible in both species. To investigate, we used ten-fold cross-validation GWAS in a large canine reference population and studied both machine learning and Bayesian regression models. Both approaches provide clinically relevant predictions with accuracies up to 69% in the dog. The performance of the machine learning models was ~10% lower than the Bayesian models. One reason for this could be that machine learning methods generally require fine-tuning of hyperparameters. The good performance of the Bayesian approach likely reflects the large number of non-contact ACL rupture risk loci and the substantial long-range LD that exists in the dog genome, which is up to 100x more extensive than in humans with low haplotype diversity within regions of high LD32. With this genetic architecture, many SNPs would be expected to be in LD with at least one risk variant, and, therefore, to have non-zero effects56. For a heritable polygenic trait and many small effect risk SNPs, a Bayesian variable selection approach typically has superior performance57. In dogs, use of ensemble prediction further increased prediction accuracy. Although preliminary, the coefficient of determination for prediction of human ACL rupture looked reasonable from GWAS summary statistic data and would be expected to improve with analysis of SNP data from individuals with accurate phenotyping. This is an important future research goal. Since surgical treatment is not disease-modifying in both species and the development of knee OA is typical5, accurate PRS prediction has the potential for high clinical impact as individuals with high genetic risk can receive personalized medical care through injury prevention screening to mitigate risk of non-contact ACL rupture and reduce disability. In dogs, genetic testing is expected to be widely adopted, particularly for high-risk breeds, such as the Rottweiler, Newfoundland, and Labrador Retriever9.
Interestingly, when we considered non-contact ACL rupture PRS prediction in a multi-ancestry dog population prediction accuracy was reduced, but was improved by admixing dogs with different ancestry in the reference population used for model training. This suggests that genetic heterogeneity exists for non-contact ACL rupture in individual dogs with differing ancestry. The principal component (PC) analysis we performed clearly shows the genetic separation of the two breeds with differing LD and MAF patterns in the two dog ancestral populations. Our analysis sheds light on the complexity of undertaking non-contact ACL rupture PRS prediction in a muti-ancestry population. These findings can be attributed to the intricate interplay of genetic and environmental factors. The genetic diversity inherent to different dog breeds, stemming from generations of selective breeding for specific traits, gives rise to unique genetic architectures. Because non-contact ACL rupture is a highly polygenic trait influenced by numerous genetic variants, the degree to which PRS values capture these small genetic effects hinges on the similarity of risk SNPs across populations. Population-specific variation in allele frequencies and genetic variants as well as differences in environmental effects contribute to these divergent outcomes. Our results in dogs model an important consideration for PRS prediction in human populations, where many PRS approaches are optimized for individuals of European ancestry potentially limiting application to other ancestral populations58.
There were several limitations to this research. The reference populations in this study consisted entirely of Labrador Retrievers and Rottweilers. Whether our findings extend to other dog breeds with differing ancestry is unclear. More extensive analysis of the genetic architecture of human non-contact ACL rupture as well as cross-species pleiotropy between humans and dogs is needed to fully identify biological pathways that are particularly enriched with non-contact ACL rupture candidate genes. While risk genes are shared between the two species, risk SNPS are not, so our perspective is that PRS prediction is most appropriately developed as an optimized approach for each species that could be enriched by knowledge of shared risk genes. Imputation of dog SNPs using whole genome sequence data could be a consideration in future work. Further expansion of the reference population of Labrador Retrievers would also be helpful to increase power of association for detection of additional small effect variants. Human biobank phenotypes are less accurate that the prospectively recruited dog reference population data for non-contact ACL rupture case/control classification. For the dog GWAS subjects recruited at UW Madison, sex, neutering, age, weight, and coat color were also recorded. The intent is to consider these covariates further in dog GWAS in future work focused on PRS prediction in veterinary medicine. However, the dog signalment covariates are not directly relevant to the cross-species analysis presented in the current study. Because we wanted to undertake a consistent analysis in humans and dogs, dog covariate phenotypes were not considered in detail in the current report.
In summary, non-contact ACL rupture should be considered a disease rather than an injury because it is a heritable highly polygenic trait with thousands of non-null variants contributing to intrinsic risk of ligament rupture in both humans and dogs that include shared risk genes in the two species. Recent findings continue to challenge the existing dogma that human non-contact ACL rupture is predominantly due to a single maneuver that catastrophically overloads the ACL10. The trait is under negative selection in both species. Highly accurate dog phenotypes could be used to enrich human biobank GWAS results as a novel approach to the identification of human candidate genes for common complex polygenic diseases. Despite the potential relevance of PRS prediction to orthopaedic disease, studies investigating its applicability are scarce. Our findings suggest that accurate PRS prediction of non-contact ACL rupture risk is an achievable research goal in both species. Clinical implementation would help identify individuals for personalized medical and physical therapy care, lifestyle modifications, and preventive measures. The spontaneous dog model also presents an opportunity for studies of novel therapy.
Methods
Labrador Retriever and Rottweiler reference population
We have complied with all relevant ethical regulations for animal use. Client-owned purebred Labrador Retrievers and Rottweilers (Canis lupus familiaris) were recruited at the University of Wisconsin–Madison UW Veterinary Care hospital through online advertising and local and national breed clubs. All procedures were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the American Veterinary Medical Association and Institutional Animal Care and Use Committee approval (Protocols V1070, V5463). A single non-contact ACL rupture diagnosed by a veterinarian was sufficient to consider the patient a case. In most cases, ACL rupture was confirmed during knee stabilization surgery. Control dogs were ≥8 years with no palpable knee laxity on orthopaedic exam and knee radiographs with no evidence of effusion or osteophytosis consistent with non-contact ACL rupture59. This cutoff was chosen because Labrador Retrievers ≥8 years have ~6% chance of developing ACL rupture60. For Rottweilers, the age cutoff was ≥5 years as the disease often develops in younger Rottweilers. Follow-up contact with owners of control dogs was used to help ensure accurate phenotyping. If a control dog developed non-contact ACL rupture, its phenotype was updated.
DNA was isolated from a saliva swab or blood obtained by venipuncture using Genotek reagents (Genotek, Ottawa, Canada) or the DNeasy kit (Qiagen, Valencia, CA, United States), respectively. DNA quantity and purity were assessed using a Qubit 4 Fluorometer (Thermo Scientific, Waltham, MA, United States) and a Nanodrop LiteSpectrophotometer (Thermo Scientific, Waltham, MA, United States). Genotyping was performed using the Illumina CanineHD BeadChip (~172,000 SNPs, or ~230,000 canfam3.1 SNPs for more recent samples) or the Thermofisher CanineHD Array (~710,000 canfam3.1 SNPs). We assembled a metadata set of non-contact ACL rupture case and control dogs. The first one was the Wisconsin dataset contained 719 dogs (326 cases, 383 controls) and the second was provided by Cornell University that used the same genotyping platform61 to increase sample size by 287 Labrador Retriever dogs (114 cases, 173 controls). For these dogs, DNA samples were obtained from blood samples collected in accordance with Cornell University animal care and use guidelines (IACUC #2005-0151 and #2011-0061)61. The final dataset consisted of genotyping data and covariates for 1,006 Labrador Retrievers (440 cases, 566 controls, 490 males, 516 females) and 108 Rottweilers (83 cases, 25 controls, 40 males, 68 females). The Labrador Retrievers ranged in age from 1 to 15 years and the Rottweilers from 1 to 12 years. The proportion of cases in the Labrador Retriever population was 43.74%. Quality control on the imputed genotypic data was performed using PLINK v1.962. SNPs were removed from the dataset if they had minor allele frequency (MAF) < 0.01, SNP genotyping call rate <95%>P value < 1E-06. After quality control 142,071 SNPs remained for analysis, and 9903 SNPs were filtered out because they did not fit with Hardy–Weinberg proportions.
Human reference population
For estimating heritability and non-contact ACL rupture PRS values, we used GWAS summary statistics, which were prepared using two cohorts of individuals with European ancestry: the Kaiser Permanente Research Board (KPRB) and the UK Biobank. The KPRB cohort comprised 83,414 individuals genotyped at 670,572 SNPs. Imputation was performed to expand the genotypic information to 12,365,897 SNPs using the 1,000 Genomes Project as a reference panel. The UK Biobank cohort included 438,669 individuals, and genotype data were centrally imputed using the Haplotype Reference Consortium and UK10k + 1000GP3 reference panels. Strict quality control procedures were implemented, excluding individuals based on genotyping missingness rate, sex inconsistency, withdrawal, and non-European ancestry, resulting in the exclusion of 18.9% of subjects from the KPRB dataset and 3.1% from UK Biobank dataset. Additional quality control measures were applied to filter genetic variants, ensuring data reliability. For the KPRB subjects, ACL injury cases was identified based on clinical diagnoses captured in the Kaiser Permanente Northern California electronic health record. For the UK Biobank subjects, International Classification of Diseases, Ninth Revision (ICD-9) or Tenth Revision (ICD-10) codes were used to identify cases of ACL injury.
Heritability estimation
We used Restricted Maximum Likelihood (REML) regression analysis of case/control status with GCTA and a probit Bayesian linear mixed model as implemented in the BGLR package63 to estimate heritability on the observed scale (h2o) in the dog. We then converted h2o to the unobserved liability continuous scale (h2l). A population prevalence of 5.79%9 was used for this calculation (SI Appendix, Supplementary Methods). BayesR and BayesS were also used to estimate heritability.
Regional heritability mapping of non-contact ACL rupture
The genome was divided into genomic regions (30 SNPs) using sequential distinct windows of markers to assess the contribution from each region to non-contact ACL rupture variance (SI Appendix, Supplementary Methods, Fig. S2). We employed the GCTA tool to estimate window-based heritability using the REML (restricted maximum likelihood) method, which provides a robust statistical framework for partitioning genetic variance across genomic regions. This approach allowed for a refined understanding of the genetic architecture influencing trait variability.
Dissecting the genetic architecture of non-contact ACL and evidence of selection using BayesR and BayesS
To study the genetic architecture of non-contact ACL rupture we used BayesR, a hierarchical Bayesian mixture model of four normal distributions each with zero mean but with variance ranging from zero to 1% of total genetic variance where SNP associations are treated as random effects drawn from a normal distribution to enable an unbiased estimate of variance explained by the SNPs (SI Appendix, Supplementary Methods). We also investigated signatures of negative selection in the genetic architecture of ACL rupture by analyzing the relationship between effect size and MAF using the Bayesian mixed linear model BayesS (SI Appendix, Supplementary Methods).
Cross-species pleiotropy analysis for risk of non-contact ACL rupture
For the Labrador Retriever, a logistic linear mixed model GWAS was performed (SI Appendix, Supplementary Methods, Fig. S10), and the top 1000 SNP associations were used for gene-based analysis. GWAS summary statistics were obtained for a human ACL rupture GWAS49, and the top 1000 SNPs were selected. Risk SNPs ±50 kb flanking regions in each species were mapped to genes via the UCSC Genome Browser (https://genome.ucsc.edu). The Fisher Exact was then used to determine whether the proportion of shared genes was higher than would be expected by chance. Results were considered significant at P < 0.05. We also pooled previously reported non-contact ACL rupture risk genes with our findings and made two gene lists for humans and dogs. For human non-contact ACL rupture, there were 66 genes, while for dog ACL rupture there were 58 genes (SI Appendix, Figs. S4 and S5), of which 50 are shared in the two species (SI Appendix, Supplementary Data).
SNP selection for polygenic risk score prediction of non-contact ACL rupture risk in dogs
A logistic linear mixed model GWAS was used to determine SNP associations with non-contact ACL rupture (SI Appendix, Supplementary Methods, Fig. S10). We conducted a grid search from 1% to 10% of top GWAS SNPs for machine learning models and 1% to 30% for Bayesian models (SI Appendix, Fig. S11). All Bayesian models achieved their best performance when 30% of the SNPs were selected. This optimization to use fewer SNPs for prediction offers several benefits. Primarily, it enhances model interpretability, allowing a focus on the most informative genetic markers. This reduction in complexity can also improve the computational efficiency of the analysis. Furthermore, by limiting the number of SNPs, the model can generalize better to new datasets, such as multi-ancestry analysis, by minimizing overfitting, thereby potentially increasing the robustness and applicability of the predictive model across different populations or environments. In all models, we included sex as the covariate that could potentially influence the prediction of non-contact ACL rupture risk.
Machine learning prediction models for risk of non-contact ACL rupture in dogs
We used four different machine learning models to analyze our data: EN, LASSO, RF, and SVM. Each model was chosen for its unique strengths in handling large datasets and complex relationships, allowing us to compare their performance and identify the most effective approach for our specific application (SI Appendix, Supplementary Methods).
Bayesian regression prediction models for risk of non-contact ACL rupture in dogs
BB, BC, BL, and BRR models were fitted and compared in terms of their prediction accuracy (SI Appendix, Supplementary Methods). Analyses were carried out using a Monte Carlo Markov chain algorithm with a total of 1,000,000 iterations and burn-in of 100,000 iterations using BGLR R package63. Global convergence was checked by visual inspection of trace plots.
Accuracy of non-contact ACL rupture risk prediction in dogs
Ten-fold cross-validation GWAS was used to investigate the accuracy of non-contact ACL rupture risk prediction. The predictive performance was assessed by several metrics, including (1) AUC of the receiver operating characteristic (ROC) analysis, (2) Accuracy, a fundamental measure of a model’s overall correctness, defined as the proportion of true results (both true positives and true negatives) among the total number of cases examined. Accuracy = (TP + TN)/(TP + FP + TN + FN), where TP = True Positives, TN = True Negatives, FP = False Positives, and FN = False Negatives. (3) F1 Score = 2×(Precision+Recall)/(Precision×Recall), Precision is the accuracy of positive predictions formulated as Precision = TP/(TP + FP), and recall is the ability of a model to find all the relevant cases within a dataset, expressed as Recall = TP/(TP + FN), and (4) R2 on the liability scale. Together, these metrics provided a comprehensive view of the model’s effectiveness in predicting non-contact ACL rupture, enabling an informed evaluation of its predictive power and reliability. To this end, the data were randomly split into ten parts and in each run nine folds of the data were used to train the model, and the other fold was used as the test set. Sequentially, we replaced each test set with a new one and repeated the prediction for all subsets until each of the ten subsamples was used once for validation to avoid overfitting. Then the mean and standard deviation (SD) of each metric was computed for comparing scenarios.
Accuracy of non-contact ACL rupture risk prediction in humans
We used four statistical models, lassosum, sBLUP, sBayesS and sBayesR, to analyze GWAS summary statistic data. We trained PRS models on HapMap3 SNPs in all analyses that were genotyped and cataloged as part of the third phase of the International HapMap Project. The performance of each model was estimated using the PUMAS method (SI Appendix, Supplementary Methods).
Non-contact ACL rupture PRS prediction in a multi-ancestry population of dogs
To assess the genetic heterogeneity of non-contact ACL rupture in dogs, we assessed PRS predictive performance in a multi-ancestry population of Labrador Retrievers (n = 1006) and Rottweilers (n = 108), two genetically distinct dog breeds. We considered three key scenarios: (1) a single ancestry population reference (LAB→LAB), where Labrador Retrievers were used as both the reference and testing populations to establish baseline PRS predictive performance in genetically similar populations, (2) a cross-ancestry PRS prediction where Labrador Retrievers was used as the reference population to predict ACL risk in a genetically distinct Rottweilers breed (LAB→ROT), and (3) a multi-ancestry reference (multi-ancestry→ROT), where a multi-ancestry reference population consisting of 1006 Labrador Retrievers and 65 Rottweilers was employed to predict non-contact ACL rupture risk exclusively in Rottweilers. This approach allowed us to evaluate the ability of PRSs to provide population-specific and multi-ancestry predictions (SI Appendix, Supplementary Methods).
Statistics and reproducibility
The data were analyzed in R environment (version 4.4.0), or command line software programs as described in the Supplementary Methods. Sample sizes for GWAS and PRS prediction analysis are described in the methods. SNP data were filtered to retain high-quality SNPs. Statistical significance was established at a P value of less than 0.05, or by correction of this threshold for multiple comparisons. Cross-validation replicates were used for some PRS prediction analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The canine datasets generated and analyzed in this study are available in the Dryad repository64. The Dryad repository plink files contain SNPs from all dogs with separate file sets for the Labrador Retriever population and the Rottweiler population. However, the ACL rupture phenotypes in the fam file for each breed have been removed because of restrictions relating to the development of a commercial genetic screening test for non-contact ACL rupture in the dog at the University of Wisconsin–Madison. Access to the human data used in this study may be obtained by application to the KPRB via kp/org/researchbank/researchers. A subset of the GERA cohort consented for public use can be found at NIH/dbGaP: phs000674.v3.p3. Data used for the preparation of manuscript figures are available from the figshare repository together with the image montages65.
References
-
Lewis, C. M. & Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12, 44 (2020).
-
Giudicessi, J. R., Kullo, I. J. & Ackerman, M. J. Precision cardiovascular medicine: state of genetic testing. Mayo Clin. Proc. 92, 642–662 (2017).
-
Douville, N. J. et al. Use of a polygenic risk score improves prediction of myocardial injury after non-cardiac surgery. Circ. Genom. Precis. Med. 13, e002817 (2020).
-
Li, Z. et al. Polygenic risk scores have high diagnostic capacity in ankylosing spondylitis. Ann. Rheum. Dis. 80, 1168–1174 (2021).
-
Binversie, E. E. et al. Canine ACL rupture: a spontaneous large animal model of human ACL rupture. BMC Musculoskelet. Disord. 23, 116 (2022).
-
Kaynak, M., Nijman, F., van Meurs, J., Reijman, M. & Meuffels, D. E. Genetic variants, and anterior cruciate ligament rupture: a systematic review. Sports Med. 47, 1637–1650 (2017).
-
Magnusson, K., Turkiewicz, A., Hughes, V., Frobell, R. & Englund, M. High genetic contribution to anterior cruciate ligament rupture: Heritability~ 69%. Br. J. Sports Med. 55, 385–389 (2021).
-
Montalvo, A. M. et al. What’s my risk of sustaining an ACL injury while playing sports? A systematic review with meta-analysis. Br. J. Sports Med. 53, 1003–1012 (2019).
-
Witsberger, T. H., Villamil, J. A., Schultz, L. G., Hahn, A. W. & Cook, J. L. Prevalence of and risk factors for hip dysplasia and cranial cruciate ligament deficiency in dogs. J. Am. Vet. Med Assoc. 232, 1818–1824 (2008).
-
Beaulieu, M. L., Ashton-Miller, J. A. & Wojtys, E. M. Loading mechanisms of the anterior cruciate ligament. Sports Biomech. 22, 1–29 (2023).
-
Bleedorn, J. A. et al. Synovitis in dogs with stable stifle joints and incipient cranial cruciate ligament rupture: a cross‐sectional study. Vet. Surg. 40, 531–543 (2011).
-
Muir, P. et al. Contralateral cruciate survival in dogs with unilateral non-contact cranial cruciate ligament rupture. PLoS. ONE 6, e25331 (2011).
-
Paterno, M. V., Rauh, M. J., Schmitt, L. C., Ford, K. R. & Hewett, T. E. Incidence of second ACL injuries 2 years after primary ACL reconstruction and return to sport. Am. J. Sports Med. 42, 1567–1573 (2014).
-
Griffin, L. Y. et al. Noncontact anterior cruciate ligament injuries: risk factors and prevention strategies. J. Am. Acad. Orthop. Surg. 8, 141–150 (2000).
-
Lohmander, L. S., Englund, P. M., Dahl, L. L. & Roos, E. M. The long-term consequence of anterior cruciate ligament and meniscus injuries: osteoarthritis. Am. J. Sports Med. 35, 1756–1769 (2007).
-
Smith, H. C. et al. Risk factors for anterior cruciate ligament injury: a review of the literature – part 1: neuromuscular and anatomic risk. Sports Health 4, 69–78 (2012).
-
Smith, H. C. et al. Risk factors for anterior cruciate ligament injury: a review of the literature -part 2: hormonal, genetic, cognitive function, previous injury, and extrinsic risk factors. Sports Health 4, 155–161 (2012).
-
Beaulieu, M. L. et al. Loading mechanisms of the anterior cruciate ligament. Sports Biomech. 22, 1–29 (2023).
-
Beaulieu, M. L. et al. The anterior cruciate ligament can become hypertrophied in response to mechanical loading: a magnetic resonance imaging study in elite athletes. Am. J. Sports Med. 49, 2371–2378 (2021).
-
Comerford, E. J., Smith, K. & Hayashi, K. Update on the aetiopathogenesis of canine cranial cruciate ligament disease. Vet. Comp. Orthop. Traumatol. 24, 91–98 (2011).
-
Robinson, M. R., Wray, N. R. & Visscher, P. M. Explaining additional genetic variation in complex traits. Trends Genet. 30, 124–132 (2014).
-
Hasani, S., Feller, J. A. & Webster, K. E. Familial predisposition to anterior cruciate ligament injury: A systematic analysis with meta-analysis. Sports Med. 52, 2657–2668 (2022).
-
Baker, L. A. et al. Genome-wide association analysis in dogs implicates 99 loci as risk variants for anterior cruciate ligament rupture. PLoS ONE 12, e0173810 (2017).
-
Nielen, A. L., Knol, B. W., Van Hagen, M. A. & van der Gaag, I. Genetic and epidemiological investigation of a birth cohort of boxers. Tijdschr. Diergeneeskd. 128, 586–590 (2003).
-
Wilke, V. L. et al. Inheritance of rupture of the cranial cruciate ligament in Newfoundlands. J. Am. Vet. Med. Assoc. 228, 61–64 (2006).
-
Cook, S. R., Conzemius, M. G., McCue, M. E. & Ekenstedt, K. J. SNP-based heritability and genetic architecture of cranial cruciate ligament rupture in Labrador Retrievers. Anim. Genet. 51, 824–828 (2020).
-
Baker, L. A. et al. Biologically enhanced genome-wide association study provides further evidence for candidate loci and discovers novel loci that influence risk of anterior cruciate ligament rupture in the dog model. Front. Genet. 12, 593515 (2021).
-
Chen, H. et al. A genetic risk score combining ten psoriasis risk loci improves disease prediction. PLoS ONE 6, e19454 (2011).
-
Baker, L. A. et al. Bayesian and machine learning models for genomic prediction of anterior cruciate ligament rupture in the canine model. G3 10, 2619–2628 (2020).
-
Wood, A. R. et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186 (2014).
-
Lello, L. et al. Accurate genomic prediction of human height. Genetics 210, 477–497 (2018).
-
Sutter, N. B. et al. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res. 14, 2388–2396 (2004).
-
Mather, R. C. III et al. Societal and economic impact of anterior cruciate ligament tears. J. Bone Jt. Surg. 95, 1751–1759 (2013).
-
Wilke, V. L., Robinson, D. A., Evans, R. B., Rothschild, M. F. & Conzemius, M. G. Estimate of the annual economic impact of treatment of cranial cruciate ligament injury in dogs in the United States. J. Am. Vet. Med. Assoc. 227, 1604–1607 (2005).
-
Eskin, E. Discovering genes involved in disease and the mystery of missing heritability. Commun. ACM 58, 80–87 (2015).
-
Hannan, A. J. Tandem repeat polymorphisms: modulators of disease susceptibility and candidates for ‘missing heritability. Trend Genet. 26, 59–65 (2010).
-
Binversie, E. E. et al. Analysis of copy number variation in dogs implicates genomic structural variation in the development of anterior cruciate ligament rupture. PLoS ONE 15, e0244075 (2020).
-
Schoenebeck, J. J. & Ostrander, E. A. Insights into morphology and disease from the dog genome project. Ann. Rev. Cell. Dev. Biol. 30, 535–560 (2014).
-
Parker, H. G., Shearin, A. L. & Ostrander, E. A. Man’s best friend becomes biology’s best in show: genome analyses in the domestic dog. Ann. Rev. Genet. 44, 309–336 (2010).
-
Baird, A. E. G., Carter, S. D., Innes, J. F., Ollier, W. & Short, A. Genome‐wide association study identifies genomic regions of association for cruciate ligament rupture in Newfoundland dogs. Anim. Genet. 45, 542–549 (2014).
-
Baker, L. A. et al. Multivariate genome-wide association analysis identifies novel and relevant variants associated with anterior cruciate ligament rupture risk in the dog model. BMC Genet. 19, 39 (2018).
-
Binversie, E. E. et al. Across breed genetic investigation of canine hip dysplasia, elbow dysplasia, and anterior cruciate ligament rupture using whole genome sequencing. Front. Genet. 13, 913354 (2022).
-
Boyko, A. R. et al. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 8, e1000451 (2010).
-
Rimbault, M. et al. Derived variants at six genes explain nearly half of size reduction in dog breeds. Genome Res. 23, 1985–1995 (2013).
-
Wray, N. R. et al. Common disease is more complex than implied by the core gene omnigenic model. Cell 173, 1573–1580 (2018).
-
Nagamine, Y. et al. Localising loci underlying complex trait variation using regional genomic relationship mapping. PLoS ONE 7, e46501 (2012).
-
Degtyareva, A. O., Antontseva, E. V. & Merkulova, T. I. Regulatory SNPs: altered transcription factor binding sites implicated in complex traits and diseases. Int. J. Mol. Sci. 22, 6454 (2021).
-
Zeng et al. Widespread signatures of natural selection across human complex traits and functional genomic categories. Nat. Commun. 12, 1164 (2021).
-
Kim, S. et al. Genome-wide association screens for Achilles tendon and ACL tears and tendinopathy. PLoS ONE 12, e0170422 (2017).
-
Kim, S. K., Nguyen, C., Avins, A. L. & Abrams, G. D. Three genes associated with anterior and posterior cruciate ligament injury: a genome-wide association analysis. Bone Jt. Open 2, 414–421 (2021).
-
Rioux, G. et al. Gene profiling of a 3D psoriatic skin model enriched in T cells: downregulation of PTPRM promotes keratinocyte proliferation through excessive ERK1/2 signaling. Cells 11, 2904 (2022).
-
Kim, M., Morales, L. D., Jang, I. S., Cho, Y. Y. & Kim, D. J. Protein tyrosine phosphatases as potential regulators of STAT3 signaling. Int. J. Mol. Sci. 19, 2708 (2018).
-
Naraoka, T. et al. Time-dependent gene expression and immunohistochemical analysis of the injured anterior cruciate ligament. Bone Jt. Res. 1, 238–244 (2012).
-
Sha, Y., Afandi, R., Zhang, B., Yang, L. & Lv, Y. MGF E peptide pretreatment improves collagen synthesis and cell proliferation of injured human ACL fibroblasts via MEK-ERK1/2 signaling pathway. Growth Factors 35, 29–38 (2017).
-
Melnyk, M. & Gollhofer, A. Submaximal fatigue of the hamstrings impairs specific reflex components and knee stability. Knee Surg. Sports Traumatol. Arthrosc. 15, 525–532 (2007).
-
Bandeira e Sousa, M. et al. Genomic-enabled prediction in maize using kernel models with genotype × environment interaction. G3 7, 1995–2014 (2017).
-
Meher, P. K., Rustgi, S. & Kumar, A. Performance of Bayesian and BLUP alphabets for genomic prediction: analysis, comparison and results. Heredity 128, 519–530 (2022).
-
Cavazos, T. B. & Witte, J. S. Inclusion of variants discovered from diverse populations improves polygenic risk score transferability. HGG Adv. 2, 100017 (2021).
-
Chuang, C. et al. Radiographic risk factors for contralateral rupture in dogs with unilateral cranial cruciate ligament rupture. PLoS ONE 9, e106389 (2014).
-
Reif, U. & Probst, C. W. Comparison of tibial plateau angles in normal and cranial cruciate deficient stifles of Labrador retrievers. Vet. Surg. 32, 385–389 (2003).
-
Hayward, J. et al. Complex disease and phenotype mapping in the domestic dog. Nat. Commun. 7, 10460 (2016).
-
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
-
Pérez, P. & de Los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 198, 483–495 (2014).
-
Muir, P. et al. Cross-species analysis of genetic architecture and polygenic risk scores for non-contact ACL rupture in dogs and humans [Dataset]. Dryad https://doi.org/10.5061/dryad.47d7wm3ns (2024).
-
Momen, M. et al. Cross-species analysis of genetic architecture and polygenic risk scores for non-contact ACL rupture in dogs and humans [Dataset]. Figshare https://doi.org/10.6084/m9.figshare.27857730 (2024).
Acknowledgements
The authors gratefully acknowledge the support of Dr. Emily Binversie and Dr. Lauren Baker for this project. Further, the authors also gratefully acknowledge the support of Dr. Rory Todhunter of Cornell University who provided some of the Labrador Retriever SNP data that were used for this study. The authors also appreciatively acknowledge the support of Dr. Stuart Kim of Stanford University who facilitated access to the public human GWAS summary statistic data analyzed as part of this research. The authors would also like to thank the faculty, residents, and students throughout the University of Wisconsin–Madison UW Veterinary Care hospital and the many individual breeders and pet owners for their help in the recruitment of Labrador Retrievers and Rottweilers for this study and the community gift funding provided for this work. Dr. Tamara Scerpella contributed to the preparation of Fig. 1. MM received support from a National Library of Medicine training grant to the Computation and Informatics in Biology and Medicine Training Program (NLMT15M007359). SJS received support from the National Institutes of Health (K01OD019743-01A1). Funding for this project was also provided by the AKC Canine Health Foundation (www.akcchf.org) and the Melita Grunow Family Professorship in Companion Animal Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
P.M. obtained funding for the research; M.M. and P.M. designed the research; M.M., H.K., M.P., S.J.S., and P.M. performed the research; G.J.M.R., Z.Z., and Q.L. contributed to GWAS design and computation; M.M. and P.M. analyzed data; and M.M. and P.M. wrote the paper. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors of this manuscript have the following competing interests: P.M. and M.M. are named on US Patent US20160222451A1 “Method to predict heritable canine non-contact cruciate ligament rupture.” This does not alter our adherence to the journal’s policies on data sharing and materials. The remaining authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Hélène Choquet and Rosie Bunton-Stasyshyn. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Momen, M., Kearney, H.K., Patterson, M.M. et al. Cross-species analysis of genetic architecture and polygenic risk scores for non-contact ACL rupture in dogs and humans.
Commun Biol 8, 26 (2025). https://doi.org/10.1038/s42003-024-07395-9
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s42003-024-07395-9
This post was originally published on this site be sure to check out more of their content.